近日,我校化学与分子工程学院计算化学中心/工业催化研究所王海丰教授课题组在Journal of the American Chemical Society上发表了题为“Topology-Determined Structural Genes Enable Data-Driven Discovery and Intelligent Design of Potential Metal Oxides for Inert C–H Bond Activation”的研究论文,在线报道了团队在机器学习辅助催化材料结构寻优方面的最新研究成果。
对于给定化学反应,识别并预测具有高活性(低能垒)的优质催化材料拓扑结构是催化剂理性设计领域的关键科学问题之一。由于多相催化材料的结构复杂性和成分的多样性,传统的Brønsted-Evans-Polanyi(BEP)线性标度关系在预测反应能垒的有效性方面存在显著缺陷。因此,对催化剂活性位点的拓扑结构进行定量描述、并开发一种迅速而准确的方法预测反应能垒具有显著意义。
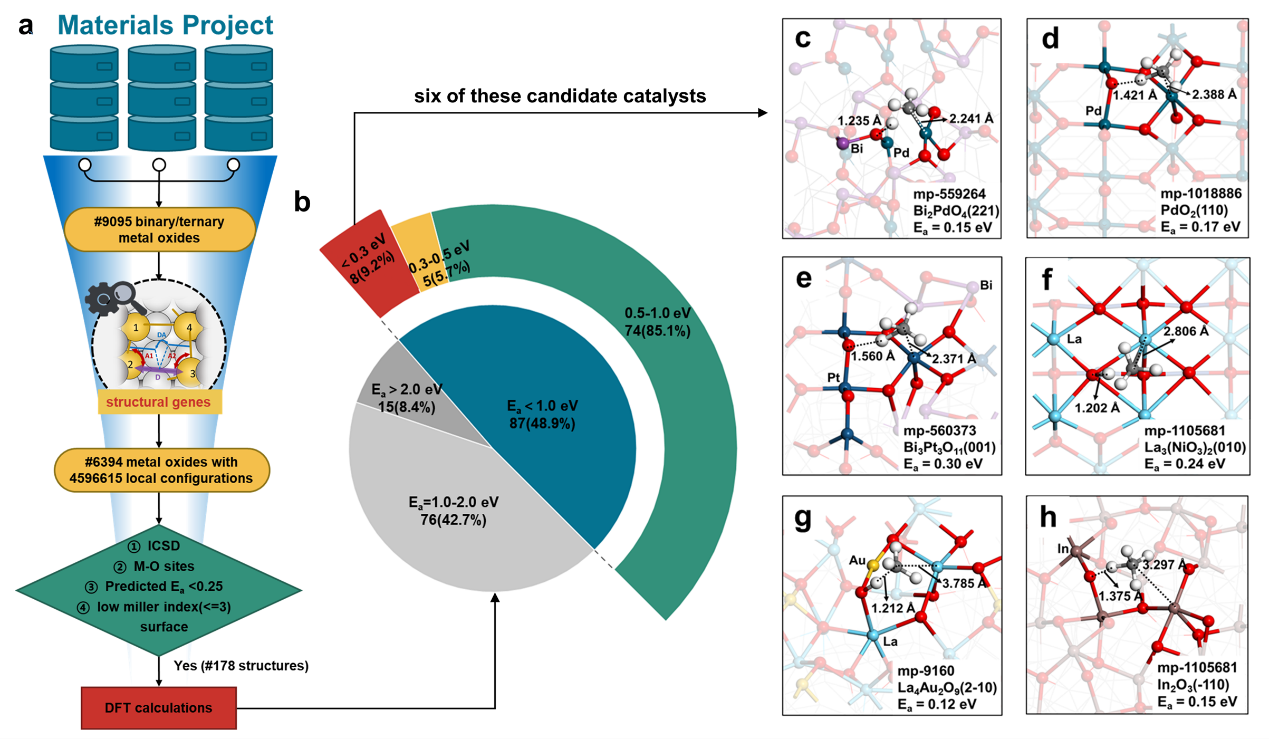
为了解决这一问题,团队以甲烷惰性C-H键的低温催化活化为应用示例,系统研究了不同类型金属氧化物上的甲烷活化过程;创新提出了一种四维度结构描述符,并建立了基于梯度提升决策树框架的机器学习模型,实现了催化活性中心结构特征的定量表达和C-H键活化能垒的预测。与传统的BEP标度关系相比,该模型的预测准确性显著提高,可以明确阐释拓扑依赖的结构-反应性关系,例如解释了金红石型IrO2(110)具有卓越甲烷活化能力的结构基础。同时,基于该机器学习模型和材料数据库,研究团队建立了自动化的催化剂高通量筛选工作流,在9095个二元和三元金属氧化物中快速筛选出了178个具有优异结构特征的候选催化剂,并最终确定了13个C-H键活化能垒低于0.5 eV的催化剂,可供进一步实验验证。该工作也为其他化学反应优质催化剂的高通量筛选提供了方法借鉴。
我校化学与分子工程学院博士研究生周川和陈琛为论文共同第一作者,王海丰教授为通讯作者。该工作得到了绿色化工和工业催化国家重点实验室、结构可控先进功能材料及其制备教育部重点实验室、国家重点研发计划和国家自然科学基金等项目的支持。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.3c06166